molamola

molamola
A Python plotting tool for Oxford Nanopore variation data. One input in, one self-contained HTML report out.
molamola picks the right plot type from its input — VCF modes are
auto-detected from the header, the karyotype mode is selected by
the --mosdepth flag:
- SV / cytogenetics report for long-read SV VCFs (Sniffles2, cuteSV, SVIM, pbsv, NanoVar).
- Per-gene phased-haplotype panels for compound-het workup from phased + VEP-annotated small-variant VCFs (WhatsHap, HiPhase).
- Karyotype coverage report for a mosdepth
regions.bed.gz(via--mosdepth) — genome-wide CN scatter + rolling-median smooth, with an optional BAF panel beneath when paired with a small-variant VCF.
Install
pip install molamola
Or via conda — note that both bioconda and conda-forge channels are needed (pycirclize lives on conda-forge):
conda create -n molamola -c bioconda -c conda-forge molamola
Quick start
molamola --vcf sample.vcf --out reports/
# or, for a coverage karyotype:
molamola --mosdepth sample.regions.bed.gz --reference hg38 --out reports/
The plot type is picked from the input (VCF header, or --mosdepth
for karyotype mode). --out is required — molamola refuses rather
than silently writing the report next to the input file. Output is
a single self-contained HTML report — figures embedded as base64,
no external assets, opens offline.
Example output
Figures below come from running molamola’s SV mode on sample MH001 (ONT LSK114 library prep, aligned-read N50 10.4 kb, median autosomal coverage 54x). The HTML report embeds both plots back-to-back; shown separately here for clarity.
Circos plot
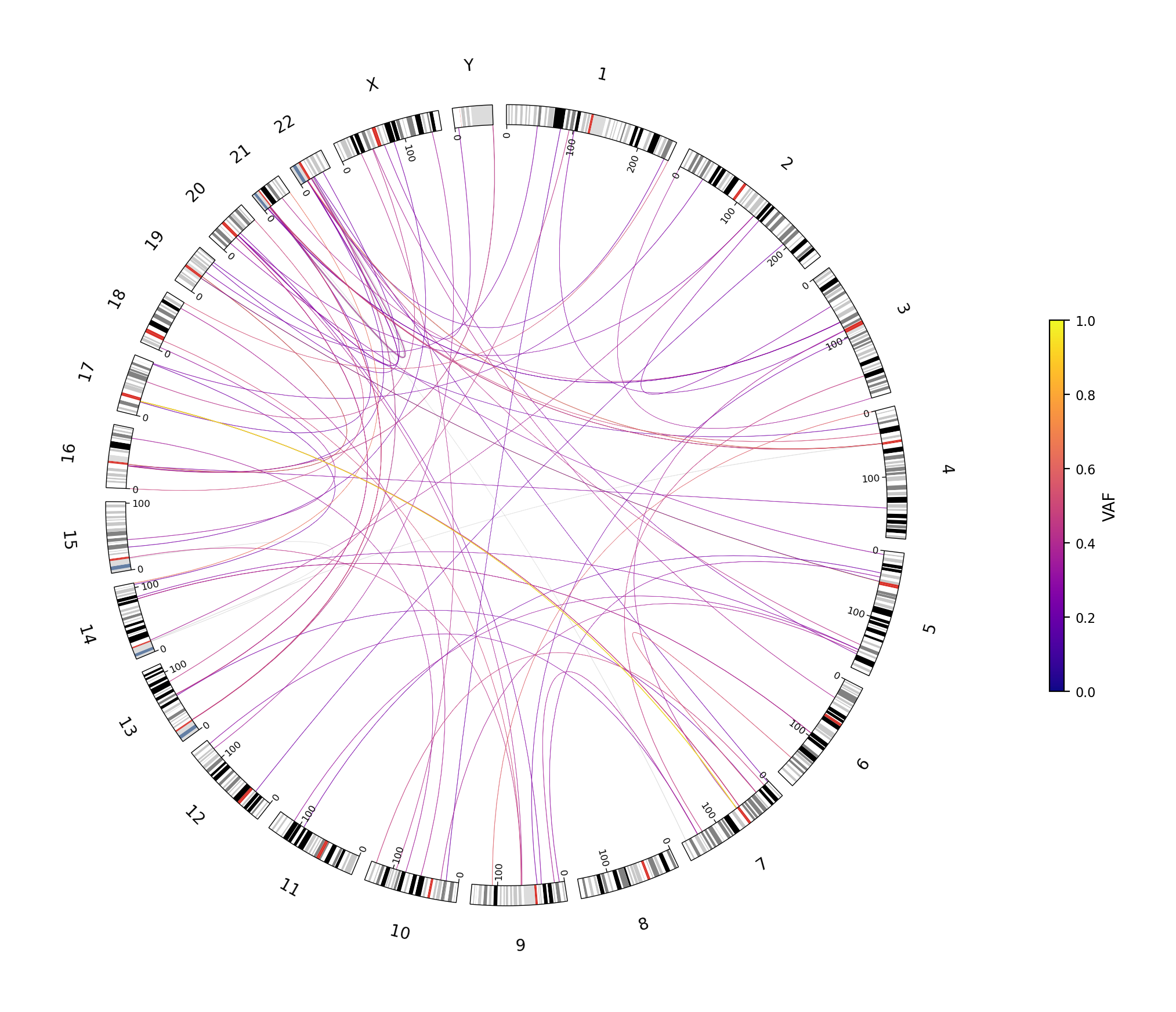
22 autosomes plus X and Y arranged around the disc, with greyscale ISCN cytobands on the rim. Each ribbon across the disc is a BND (translocation or large rearrangement); ribbon colour encodes VAF (purple = low → yellow = high, plasma colormap). At-a-glance view for inter-chromosomal events.
Linear genome map
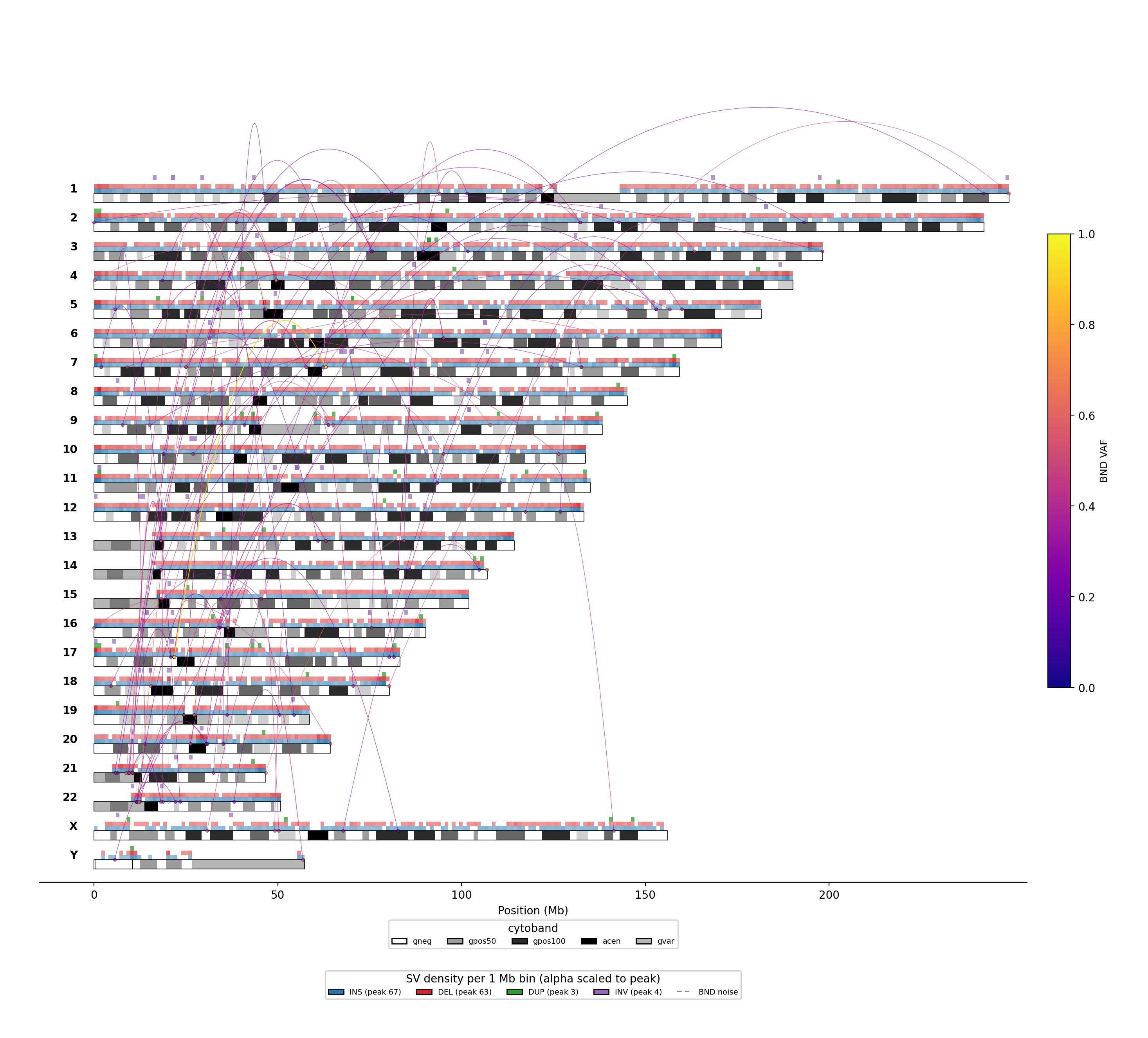
One row per chromosome (chr1 at top, chrY at bottom). Cytobands embedded inside each chromosome track. Above each track sit four 1-Mb-bin density strips — INS = blue, DEL = red, DUP = green, INV = purple — with alpha encoding per-bin event count. BND arcs hang above the tracks, colour-encoded by VAF as in the circos. Better for per-chromosome detail and density hotspots.
Compound-het example
Pending.
Preparing a phased VCF for compound-het mode
Compound-het mode needs both phasing (PS FORMAT field) and VEP
annotation (CSQ INFO field). A raw phased small-variant VCF —
e.g. straight Clair3 output — has the first but not the second,
and molamola will refuse it. Annotate with
Ensembl VEP first.
1. Download the matching VEP cache once (one-time, ~20 GB), on a machine with internet:
wget https://ftp.ensembl.org/pub/release-105/variation/indexed_vep_cache/homo_sapiens_vep_105_GRCh38.tar.gz
If your VEP isn’t 105, swap 105 for your release number (it
appears twice in the URL); the cache release must match the VEP
release exactly. Transfer to wherever you run VEP if that’s a
different machine.
2. Unpack into a stable cache directory:
mkdir -p VEP_cache && cd VEP_cache
tar -xzf ../homo_sapiens_vep_105_GRCh38.tar.gz
# creates VEP_cache/homo_sapiens/105_GRCh38/
3. Run VEP fully offline, with --canonical --symbol --pick
so the CSQ shape matches what molamola consumes:
vep --input_file sample.phased.vcf \
--output_file sample.phased.vep.vcf \
--vcf --offline \
--cache --dir_cache /path/to/VEP_cache \
--assembly GRCh38 \
--fasta /path/to/hg38.fa \
--canonical --symbol --pick \
--force_overwrite
Then molamola --vcf sample.phased.vep.vcf --out reports/ picks
it up as compound-het mode.
Notes on VEP. VEP is third-party software (Ensembl); molamola
does not bundle or wrap it. The cache release and VEP binary
release must match exactly — a mismatch leads to silent
mis-annotation rather than a clean error. Compound-het mode reads
VEP’s Consequence, SYMBOL, and CANONICAL fields as-is; any
quirks of a particular VEP build are inherited. --pick reduces
multi-transcript CSQ entries to one per variant.
Bundled references
molamola ships its own reference data inside molamola/data/:
cytoBand.txt.gz(hg38) andcytoBand.t2t.txt.gz(T2T-CHM13v2.0) — UCSC cytoband annotations, used for the SV ideogram tracks.canonical_exons.hg38.tsv.gz— MANE Select v1.x canonical transcripts and exon coordinates, used for the compound-het exon track.clinvar.hg38.tsv.xz— molamola’s reduced ClinVar TSV (chrom, pos, ref, alt, significance bucket; xz-compressed). The release date of the bundled snapshot is logged in each HTML report’s run-metadata.
Bundled-only by design: molamola does not auto-download or look up
online. Override with --clinvar PATH or
--canonical-exons PATH if you want a fresher snapshot. The two
reduced TSVs are reproducibly regeneratable from public sources
via scripts/derive_canonical_exons.py and
scripts/derive_clinvar_for_molamola.py in the repo.
Documentation
- CLI reference — every flag, with defaults and meanings.
- Examples — worked commands for each plot type.
- Filters — noise heuristics, focus windows, ClinVar gating.
- Output spec — what’s in the HTML; how figures are encoded.
Source
- Repo: github.com/martinandclaude/molamola
- PyPI: pypi.org/project/molamola
- Changelog: CHANGELOG.md
- Issues: github.com/martinandclaude/molamola/issues